ASH20 CAR-T cell therapy Preview
The king kahuna of abstract volume often gives a very good idea of the current trend or focus in any given year at ASH meetings.

Cascade of choices in the CAR T cell niche
In the past the conference has been dominated with SCT approaches, then various chemotherapies or near chemo-like agents such as Aurora kinases or Polo-like kinases with the similar toxicity profiles, then targeted therapies followed by antibodies and bispecifics.
This year chimeric antigen receptor (CAR) T cell therapies may well finally have come of age with nearly 500 abstracts to peruse across a wide gamut of companies, targets, constructs, and hematologic tumour types.
We have certainly moved on in terms of new data, not only from second generation CARs, but also simple clinical retelling of the top line data – now there’s many more subtleties and nuances to consider in terms of construct development, biomarkers, and even how various subsets respond differently depending on the underlying biology.
Figuring out how to define these key populations and better tailor therapy to the underlying biology has finally come of age in this niche, with quite a few important talks to watch out for during the upcoming meeting…
To learn more from our oncology analysis and get a heads up on the latest insights and commentary pertaining to the ASH meeting — including our third meeting Preview of 2020, subscribers can log-in or you can click to gain access to BSB Premium Content.
This content is restricted to subscribers
“Creativity is the power to reject the past, to change the status quo, and to seek new potential. Simply put, aside from using one’s imagination – perhaps more importantly – creativity is the power to act.”
~ Ai Weiwei, Chinese contemporary artist
Today’s quote is just as apt for new developments in cell therapy and cancer research as it is for the arts – we cannot stand still and live in the past, we must keep moving forward and experimenting with freedom to do so. Innovation and creativity keep us all alive. This is the first year in a while I have noticed a larger than usual cascade of fresh ideas in either new, more nuanced CAR-T product designs or creativity in the various ‘deeper dive’ analyses looking at differences in responders and non-responders, for example.
Background
It’s now eight years on from when Novartis and the University of Pennsylvania did their deal for the first commercial collaboration of a chimeric antigen receptor (CAR) T cell therapy in 2012 – then codenamed CART19.

Dr Stephan Grupp (CHOP) at ASH
For some context, consider at ASH12 there was an inevitable raft of early phase 1 data, which produced a rather exciting meeting for cell therapists… Dr Stephan Grupp (right) presented the updated results in kids with B-ALL from CHOP, with adult data (plus the update on the first child who received a CAR-T cell therapy Emily Whitehead) in CLL/ALL also coming from Dr David Porter, plus T cell persistence data from Dr Michael Kalos.
While the Penn/Novartis CAR had a 4-1BB co-stim domain, Dr Marco Davila unveiled the MSK data using their CD19 CD28 CAR in adults and Dr Daniel Lee (NCI) gave an update on the progress with their CD19 CD28 CAR.
There was also preclinical data on another CD19 CD28 CAR from the Michael Hudecek/Stan Riddell group at Fred Hutch in Seattle.
Even back then, the Penn/MSK/NCI groups were slightly further ahead than Fred Hutch, so it is not surprising they and Juno/BMS are finally close to market now, never mind the later trials and tribulations experienced by the MSK/Juno CARs. Another point to note is that not all of those early CARs made it to market – there were also some unfortunate casualties along the way.
Still, not everything CAR-related was entirely CD19 focused that year… Drs Stephen Forman and Michael Jensen explored the first CD123 CAR in AML models, for example.
In addition, the folks at the NCI reported on preclinical results from their CD22 CAR in mice. This year we get to see Stanford showcase their modified and revised CD22 CAR approach in patients, which finally led to some durable responses:
“To date, 3 of 3 heavily treated adult patients with LBCL whose disease relapsed after prior CAR19 have each achieved CR durable to at least 6 months. All adult ALL patients have achieved CR following CAR22, with some early relapses observed.”
Whether sequencing a CD19 then CD22 CARs will be a better strategy than a dual bispecific CD19/CD22 CAR such as the one from Saar Gill and Penn present viz preclinical data this year remains to be determined.
When these CD22-based constructs appeared seemingly out of nowhere in the clinic a few years later, eager students who follow CAR-T history will always remember the initial data from 2012 – we’ve clearly come a long way since then. To put the scale of the growth of CAR T cell therapies into context, there were less than 20 CAR abstracts in 2012, while this year there were… over 400, demonstrating a tremendous growth in less than a decade!
Now, we don’t plan to review all of this year’s crop as it would be a pointless exercise but we can learn a lot from the highlights and – sadly – some lowlights to review and discuss.
Before we get started on this year’s crop of abstracts, I’d like to draw your attention to a couple of important and highly relevant papers first:
i) Potential for targeting two inhibitory checkpoint targets with a CAR?
There’s a brand new preprint from researchers at the South Korean biotech Curocell demonstrating dual PD1 and TIGIT downregulation enhances CAR-T performance:
“Simultaneous, cell-intrinsic downregulation of PD–1 and TIGIT enhances the effector function of CD19-targeting CAR T cells and promotes an early-memory phenotype.”
They have several agents in preclinical development against CD19, CD22, and BCMA, which have various tweaks in the design built in, with the aim of addressing the hostile tumour microenviornment. Just as we saw bluebird tweak their manufacturing process with bb21217 to include a PI3K inhibitor (bb-207) with the aim of improving persistence, so too are other companies exploring ways to optimise and enhance their constructs. Some will succeed, others will fall short when translated into the clinic.
Comment: There’s no doubt both the PD–1 and TIGIT targets can be engineered out with modern gene editing tools, so it could be worth trying to see if there are improvements over the inconsistent data seen with addressing PD1 alone.
ii) Finding biomarkers of early response to CD19 CARs
There’s a new paper just out in Blood Advances from Dr Frederick Locke and colleagues looking at (amongst other things) cell populations at baseline which might predict responders and improved outcomes for patients with R/R NHL on axi-cel. Specifcally, they discovered something quite intriguing:
“Axi-cel durable responses were associated with low baseline tumor burden, low systemic inflammation, and high product CCR7+ CD45RA+ T cells.”

Dr Mackall at CART2020 in Sitges
But that’s not all – recall the recent discussion from Dr Crystal Mackall (right) at the Yale IO conference, where she showed how a population of CD57+ T cells at day 7 were associated with improved survival at 6 months, again in R/R DLBCL patients treated with axi-cel.
This begs the question – are these two cell populations congruent or incongruent – and what can we learn from the findings?
It turns out they are very congruent – Dr Mackall’s post doc, Dr Zinaida Good kindly responded to my query about the different cell populations in the two analyses and what they mean:
“We believe their findings – that more CCR7+ CD45RA+ T cells in axi-cel product associated with good outcome – are consistent with other reports showing that more quiescent / memory signature in CAR T-cell products are associated with better patient outcomes.
Please note that our study is focused on CAR T-cell subsets observed on day 7 following infusion. It is likely that Tscm / Tmem cell in product (including CCR7+ CD45RA+ cells) went through a proliferative burst and became CD57+ (and mostly CD45RO+) by day 7.
Thus, our findings do not contradict and are consistent with published work.”
This is very cool indeed! Based on these data we should soon be able to predict outcomes either from the quality of the starting T cell product harvested from patients or specific cell populations at Day 7 and make therapeutic adjustments accordingly.
It should be noted that despite the original idea from many observers that CAR-T cell therapy would be a one-and-done curative therapy, thereby justifying the high sticker price, in reality, for many this aspiration has not proven to be the case from the results seen to date.
If you look at the alternative concept – as a bridge to transplant – originally propounded by MSK and GOSH, this approach may have some legs with allogeneic rather than autologous products, and here’s why:
- Off-the-shelf products will be much cheaper to produce than autologous, personalised approaches
- This means more than one infusion can be given – on an as needed basis – much like we might do with antibody infusions
- Emerging biomarkers of response (e.g. at Day 7) may help us decide those most at risk of early relapse
- High risk patients could be identified early and go to SCT before they experience relapse
Let’s roll with the ASH20 peek at keep CAR abstracts. Rather than autologous CD19 CARs, we’re going to begin from an entirely different point:
1. Allogeneic CAR-T cell updates
This brings us on to an important topic… If we want to improve on the starting product of CAR-T cell therapies then one way to achieve this might be to use healthy donors – a number of companies are active in this space, including Cellectis, Allogene, Precision Biosciences, and Fate Therapeutics, for example. There are a few abstracts of interest in the allogeneic niche at ASH20:
a) ALLO–715 – Allogene
This is the Cellectis BCMA targeted CAR-T cell therapy acquired as part of the Pfizer-Servier deal in 2018 and is now undergoing clinical evaluation in multiple myeloma.
“ALLO–715 is a genetically modified anti-BCMA AlloCAR T cell product in which the TCR alpha constant gene is disrupted to reduce the risk of graft-versus-host disease (GvHD) and the CD52 gene is disrupted with Talen® technology to permit the use of ALLO–647, an anti-CD52 mAb, for selective and prolonged host lymphodepletion.”
In other words, the TCR knockout reduces the risk of GvHD, while CD52 blockade is thought to enhance the GvL effect. What happens in clinical practice?

Dr Sham Mailankody (MSK)
Dr Sham Mailankody (MSK, right) is presenting an update on the ongoing phase 1 trial in R/R MM. The abstract is presumably a placeholder. 19 patients were enrolled with 15 receiving therapy across 3 dose levels, all but one had a prior autologous SCT. The majority (52.6%, 10/19) of patients had high risk cytogenetics and 26.3% (5/19) had extramedullary disease.
Overall, they generated an activity rate of 33%, with initial activity being more robust at the third dose level:
“A higher dose of ALLO–715 (DL3) was associated with greater anti-cancer activity with 3/5 patients responding per IMWG (60%). In patients who received DL3 FCA, 2/3 responded (1 sCR and 1 VGPR, Table 1).
All DL3 patients who responded experienced at least a VGPR and achieved MRD negative status by local MRD testing. All responses were initially observed at day 14. Four (80%) out of the 5 responders were still in response at the time of the data cutoff.”
Where FCA is fludarabine, cyclosphamide, plus ALLO–715.
We previously covered Allogene at ASCO20 – a lot of folks were excited by the ALLO–715, but we were not and went so far as to make it one of our negatives in the Winners & Losers in the post meeting review. We explained why in this post. Note at least one KOL went so far to express a concern then – neither they nor I are keen on the CD52 lymphodepletion with memories of alemtuzumab (Campath) side effects ringing in our ears:
“It’s almost a lock that a patient will die on study at some point from an infection.”
Guess what we learn from the ASH abstract? A grade 5 event (death) has been reported:
“The fourth was a Grade 5 episode that occurred on day 8 post-ALLO–715 infusion in a rapidly progressing, refractory myeloma pt who, on day 1, developed a non-neutropenic fever and multifocal pneumonia with negative blood and sputum cultures. The patient progressed to respiratory failure and only comfort care was pursued. This death was considered related to conditioning.”
Comment: Sadly the KOL warning didn’t take long to materialise. In all fairness, infectious complications also occur with advanced myeloma, so adding in the CD52 lymphodepletion feature only adds to the risk of such events occurring. I got a lot of stick from readers annoyed at Allogene not being in the ‘winner’ category but time and events have only supported the initial risk assessment. At this point, there is no point in discussing the activity since updated data will likely be presented in the virtual meeting when more granular details will be available relating to the patient population and prior therapies.
b) CYAD–211 – Celyad
The Belgian biotech have been on our radar for a little while now. They are developing both allogeneic and autologous cell therapy products against targets suach as NKG2DL and BCMA. The former is the subject of a recent collaboration with Merck to explore CYAD–101 with pembrolizumab in mCRC after FOLFIRI chemotherapy in a phase 1b study, KEYNOTE-B79.
An allogeneic BCMA CAR product is being showcased at ASH. It differs from the Allogene/Cellectis product in two main ways:
- Rather than directly knockout the TCR with gene editing, this construct exploits short hairpin RNA (shRNA) interference technology to downregulate TCR expression to reduce the risk of GvHD.
- There’s no CD52 KO or lymphodepletion, reducing the risk of additional infectious complications.
Terry Fry’s group are presenting the preclinical data on what is essentially the first non-gene edited allogeneic CAR T-cell product based on shRNA technology. A phase 1 trial codenamed IMMUNICY–1 is currently enrolling patients.
Comments: This is an intriguing development I think is well worth watching out for.
2. Myeloma CAR-T cell therapy trial readouts of interest
In the past, various trials in pediatric ALL and adult R/R NHL have been the main focus of CAR T cell therapy attention, leading to the approval of tiso-cel, axi-cel, and more recently, brex-cel. None of these are included in this year’s Preview because we wish to focus on pre-approval topics and products.
This year, for example, there is a huge raft of myeloma data, including some update on next generation agents, plus a bunch of early trials with different targets in other tumour types to explore, as well as allogeneic CARs to think about.
Here are some of the myeloma talks we will be following and will be writing more during our daily coverage as the new or updated data are revealed – most of these abstracts are placeholders and may have a later cut-off for the presentations. So in no particular order:
- CRB 401 and CRB 402 – A lot of folks were no doubt expecting ide-cel (and also liso-cel) to have been approved by now, but the pandemic has thrown somewhat of a spanner in the works in this direction in terms of FDA inspections etc. Still, we will have updates on the two phase 1 trials for ide-cel and bb21217 (with the PI3Ki added) in 62 and 46 patients, respectively.
Comment: The latter particularly interests me because BMS will be sharing biomarker data on the correlation of expansion and duration of response with T cell phenotypes.
- CARTITUDE–1 – JNJ–4528 is now known as cilta-cel. Recall we saw 100% ORR from 29 patients at ASH19, now there’s more robust data from Dr Deepu Madduri, who is presenting an update on 97 patients – ORR 95%, mPFS not reached, CRS 95%, NT 21%.
- EVOLVE – Orva-cel (JCARH125) also has a preliminary high ORR of 92%, as per data at ASCO20 from Dr Sham Mailankody. This is the next generation CAR engineered to have more central memory phenotype, using a different tweak in their manufacturing approach than in the bluebird/BMS product, bb21217, which added PI3K in to boost persistence.
Take a look at the marker data provided in the new ASH20 abstract – the central memory phenotype (CCR7+ CD45RA-) is high, but so too are the naive CD8 T cells (CCR7+ CD45RA+):
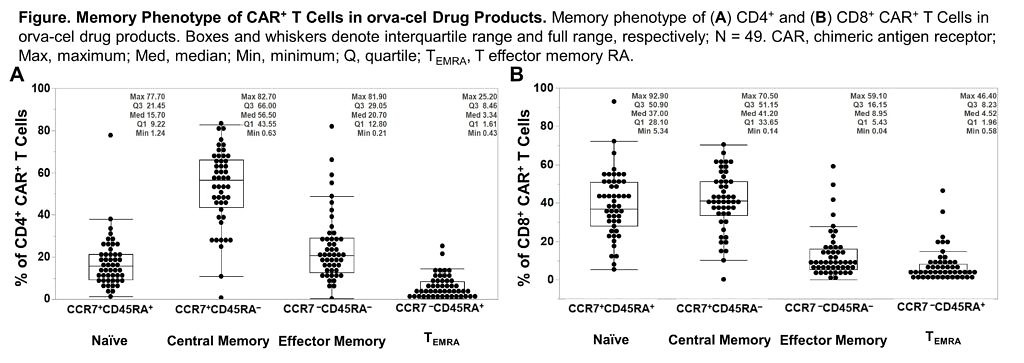
Source: Colonna et al., ASH20
Comment: The naive CD8 T cell population is the subset in Dr Locke’s axi-cel paper mentioned earlier, which were found to be associated with durable responses. What will be intriguing to see here is how the two populations play out in terms of long term outcomes – are both needed at higher levels to really boost durable activity or just the former? What additional impact will the memory phenotype have? Time will tell.
- LUMMICAR 1 and 2 – Not all the cool CARs from the US… A Chinese biotech, CARsgen Therapeutics, is developing CT053, a next generation fully human BCMA-directed autologous CAR with a high binding affinity. They have some impressive initial results from a small cohort of Chinese patients akin to what we originally saw from Legend Therapeutics:
“At the data cutoff, among 12 subjects with at least 4 weeks of efficacy assessment, a 100% ORR was observed, with 4 stringent complete responses (sCR), 1 CR, 3 very good partial responses and 4 partial responses. All 5 subjects with CR/sCR were minimal residual disease (MRD)-negative at the 10 5 sensitivity level.”
These are small (actually tiny) numbers, however, so avoid extrapolating the findings to the broader universe because a few more patients with poorer responses can shift the ORR in a downwards direction very quickly! Meanwhile the second trial is enrolling patients in the US, which is being presented by Dr Shaji Kumar (Mayo), suggesting almost identical results so far:
“At the data cutoff, 10 subjects were evaluable for at least two months of efficacy assessment with a median follow-up of 4.5 months (range 2–8). A 100% ORR was observed, with 2 stringent complete responses, 2 CRs, 1 very good partial response, and 5 partial responses. Of the 12 subjects with evaluable bone marrow samples, 11 were MRD-negative at the 10–5 sensitivity level.”
Comment: Of course, it’s still very early days yet for both trials and we need to see what happens with many more patients and much longer follow-up, but an encouraging start to date all the same.
In terms of other relevant myeloma targets – we mentioned GPCR5D in the bispecific ASH Preview and while there is an advanced dual CARs against this target (with BCMA) in preclinical development from Drs Renier Brentjens and Eric Smith at MSK (see: there’s really nice accompanying editorial from Stan Riddell), they have yet to enter clinical trials (at least in the US), so we will have to wait a little longer for some comparisons on this front to see if dual targeting reduces the immune escape and early relapse seen in some myeloma patients.
3. Early stage CARs / novel targets
This is a polyglot group illustrating how diversified CAR constructs are these days. They fall into a number of different categories – enhancement in manufacturing, innovative construct developments and even novel targets so let’s take a look at a few examples of the ilk (in alphabetical order)…
a) ArcellX

Dr David Hilbert, Source Arcellx
We talked to the folks at Arcellx (right) about the exciting and elegant science behind their out-of-the box approach to CAR-T cell therapy design earlier this year. For those of you who would like to catch up on the technical underpinnings behind the engineering using their creative Tag system, it really is quite intriguing and innovative.
At that time, they hoped to have data at ASH and here we are with the first one in their pipeline (ARC–101) with initial clinical data. Instead of the standard scFv format adopted by first and second generation CARs, they took a different approach with CART-ddBCMA. It encodes a non-scFv, synthetic binding domain targeting BCMA with a 4–1BB co-stimulatory motif and CD3-zeta T cell activation domain:
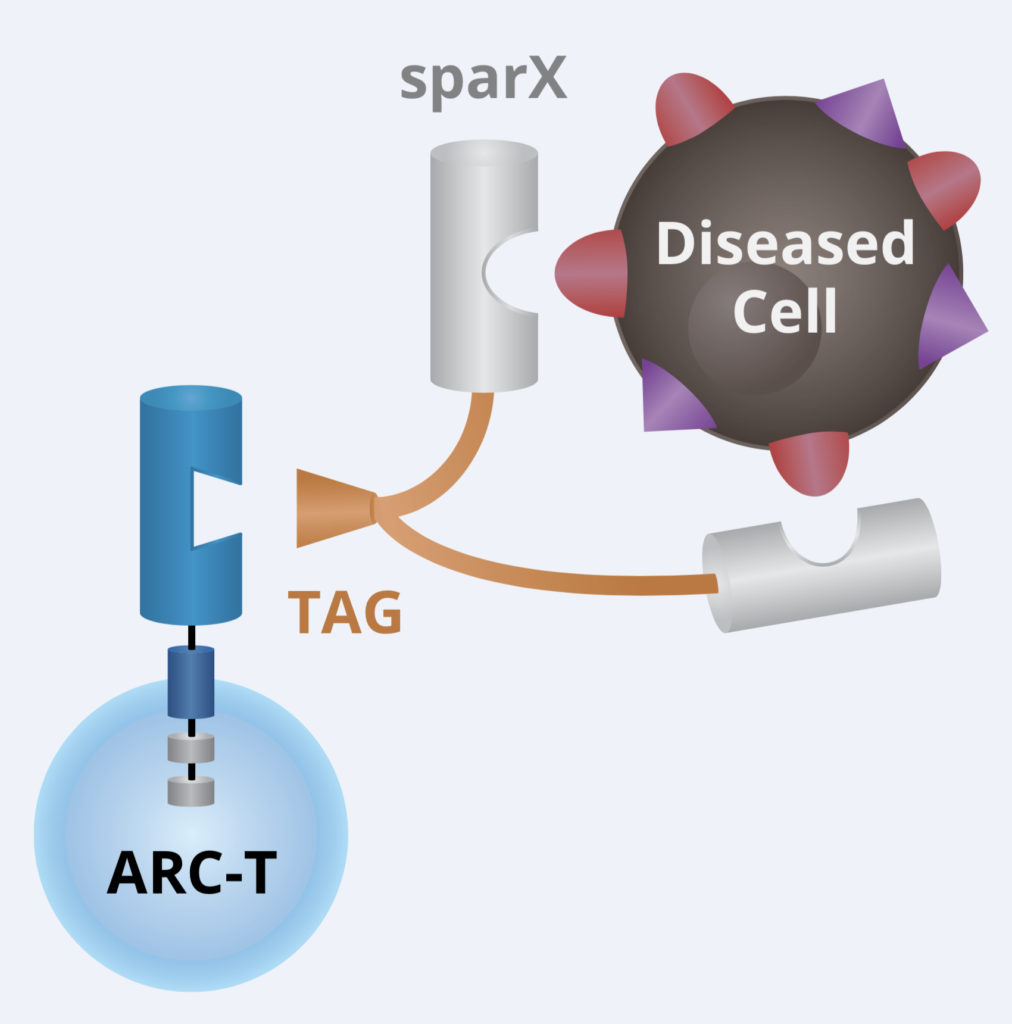
Source: Arcellx
They decided to go after BCMA as their first target. The abstract reports on the first 4 of 12 planned patients, with a median follow-up of 100 days. All were elderly subjects who did not initially receive autologous transplant following induction therapy. Transplant ineligible patients have long been a challenging group to manage for multiple myeloma doctors, with Vd or Rd being the typical treatment given in this setting for the longest time until the RVd triplet started to get some traction in those with appropriate insurance.
In all, 3 people were evaluable for initial safety and clinical response with the other pending assessment, and so might possibly be included in the actual presentation by Dr Matthew Frigault.
These were heavily pre-treated myeloma patients – all four previously received Bor/Len/Car/Pom/Dara at some point and two were penta-refractory. Three patients had high tumour burden, with 95, 95, and 70% bone marrow plasma cells pre-infusion, respectively.
What of the initial safety and activity results?
“Three subjects developed Grade 2 CRS and 1 subject developed Grade 2 ICANS. These adverse effects resolved quickly after intervention; 3 subjects received tocilizumab and 2 received steroids (dexamethasone). All 3 evaluable subjects have demonstrated clinical response per IMWG criteria: currently 1 sCR (MRD–10–4), 1 sCR, 1 sCR (MRD–10–6).”
Comment: This is an encouraging start thus far with no significant concerns such as grade 3 CRS or neurotoxicity of note. We now need to see how well ARC–101 does when administered to more patients and with longer follow-up to determine how robust the effects are.
b) MB–106 – Mustang Bio
Mustang’s MB–106 is a third generation CD20 CAR T cell therapy (MB–106) for the treatment of patients with R/R B cell NHL. You can find out more about the company and their approach from the interview with their c-suite execs here.
This is an interesting development because the abstract from the Seattle researchers highlights some of the trials and tribulations associated with CAR-T cell therapy development – it isn’t always a bed of roses, despite how the mainstream media write this niche up with garish headlines.
So 12 patients were enrolled with 11 treated with MB–106:
“The first 7 patients (3 FL, 3 MCL, 1 hairy cell variant) were treated using the original cell manufacturing method with separate culturing of CD4+ and CD8+ cells (‘original process’).”
Four of the 7 received cyclophosphamide and the other three Flu/Cy as lymphodepletion. Was there any CRS or neurotoxicity observed?
“There was no evidence of ICANS in those 7 patients and only 1 patient developed CRS (grade 3 – unexplained elevated alkaline phosphatase (ALP) in the setting of fever).”
How about activity?
“Responses at 4 weeks for patients treated with the “original process” included stable disease (SD) in 4 and progressive disease (PD) in 3 patients.”
Rather modest results overall, one must admit., but remember these would be patients who would have received prior rituximab at some point in the past. Is the problem refractoriness to CD20 or manufacturing? It turns out they looked more closely at the latter:
“Given the challenges in meeting the protocol requirements for CD4+ and CD8+ specific doses, poor CAR-T expansion, and lack of clinical responses, enrollment was placed on a hold and cell manufacturing process underwent a major revision which included changing to a combined culture of CD4+ and CD8+ cells, among other modifications (‘modified process’).”
These changes in the manufacturing process led to substantially better performance of MB–106 in the next four patients evaluated:
“Target cell doses were achieved in all 4 patients and treatment was tolerated with no occurrence of CRS (any grade) or ICANS (any grade). Responses were observed at both dose levels and included complete remissions (CRs) in 2 FL patients (1 treated at DL1 and 1 at DL2), partial remission (PR) in a MCL pt treated at DL2 and PD in a FL patient who was treated at DL1.
Robust CAR-T expansion was seen in 3 of the 4 patients, with peak blood levels of CAR+ T cells of 161, 5.5, and 401 CAR+ cells/µl, corresponding to 80%, 23%, and 72% of all CD8+ cells.”
Success! The trial continued on with the revised manufacturing process, enrollment is ongoing at higher dose levels is currently ongoing and additional data will be included in the ASH presentation.
Comment: With all the intense competition in the CD20 niche from other next generation CARs (e.g. Precision and several others), engineered toxin bodies (Molecular Templates), not to forget numerous CD20 T cell bispecific engagers from Roche, Genmab, Regeneron and others, Mustang are to be credited with going back to basics and figuring out how to improve on the quality of the starting product in order to enhance outcomes for patients. I look forward to following this development further.
c) P-BCMA–101 – Poseida Therapeutics
This is an autologous CAR targeting BCMA and manufactured using what the company call a novel transposon-based system called ‘piggyBac’ (see below), which is a non-viral system based on human centryins rather than the standard scFv backbone designed to increase efficacy while minimising toxicity. The heart of this approach is the product has a high percentage of stem cell memory T cells, presumably to improve persistence and reduce early relapse.
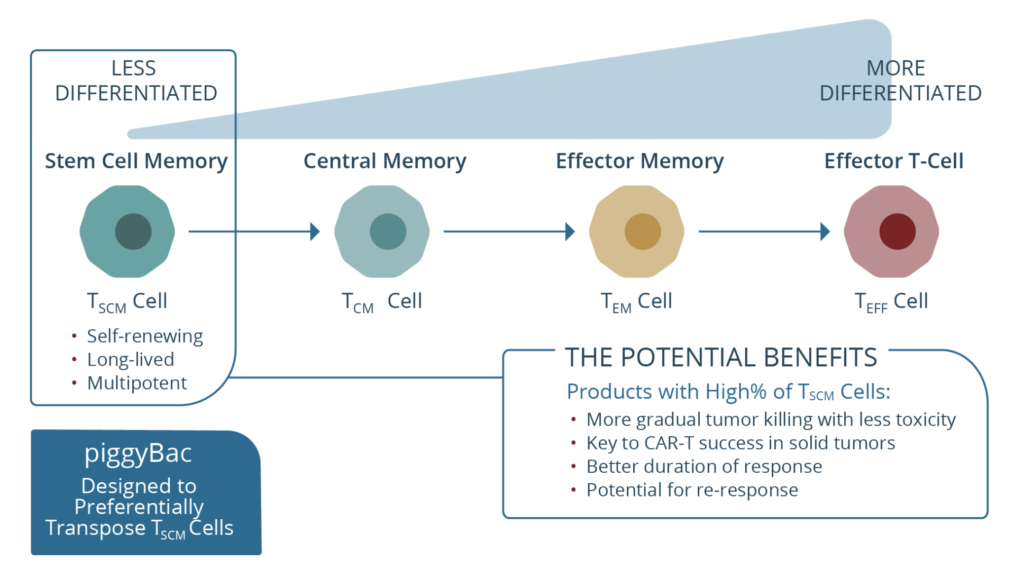
Source: Poseida Therapeutics
I’ve always been a bit sceptical of this particular approach, not because of the Tscm idea for improving central memory phenotype, but rather because I’ve yet to see any compelling activity to make me jump up and down with joy. Sure one can reduce costs, toxicities, and even have the convenience of outpatient administration, yet what matters most in oncology is outcomes and efficacy.
The abstract is a bit mixed overall, so we run the gamut from initial results in 43 patients…
“The overall response rate (ORR) for evaluable subjects (n=34) treated with single administration during the initial dose escalation was 57%.”
I know this is phase 1 dose escalation, but compare this underwhelming number to the excellent activity seen with cilta-cel or orva-cel and other second generation CARs at the same stage of development and it’s kind of meh at best. Then we get the unexpected change in tack with a ‘let’s mix things up a bit and confuse everyone’ strategy after not seeing a clear dose-response effect with lower doses looking a bit better:
“Additional cohorts were implemented focusing on the lower end of the dose range using product from the modified manufacturing process. Four patients were subsequently treated with cyclic administration, rituximab, lenalidomide or single administration at the lowest dose level with this manufacturing process (all treated with P-BCMA–101 within ~2 months prior to the data cut-off date), and thus far all have rapidly responded (100% ORR) and all responses are ongoing.”
Comment: Basically then, if at first you don’t succeed just throw the kitchen sink at the problem to boost the activity – except we have no idea what effect is due to the CD20 antibody, IMiD or CAR… and whether or not the myeloma patients were truly Revlimid-refractory. This kind of dabbling does no one any favours, frankly. There was a shadow over the company recently with their P-PSMA–101 CAR in CRPC until the clinical hold (for a death due to liver failure) was recently lifted with protocol modifications. There’s no evidence the issue was specifically due to the PiggyBac system and the PSMA target must be a stronger possibility in that particular case. Meanwhile, a decision on the RP2D is expected early 2021.
d) STK-009 OrthoCAR — Synthekine
Not all the new or unique developments with novel CAR’s have come from big pharmas. An early biotech one which looks intriguing is from Sythekine, a Stanford spinoff based on the work of Dr Christopher Garcia, the scientific founder.
Dr Rizwan Romée’s lab at Dana Farber have been extending their knowledge with cytokines such as IL-15 to boost NK cell activation to explore IL-2 and T cells.
Their latest abstract is entitled, “OrthoCARs: Engineered human IL-2/IL-2Rb orthogonal pairs selectively enhance CAR T cell antitumor efficacy”:
“To facilitate selective ex vivo and in vivo expansion of engineered T cells we have developed a human orthogonal (ortho) ligand/receptor system consisting of a pegylated, IL-2 mutein (STK-009) that does not significantly activate the wild type receptor and a mutated IL-2 Receptor Beta (orthoIL-2Rβ) that does not significantly respond to its native ligand, wild type IL-2. This system enables in vivo IL-2 signaling in engineered cells that express the orthoIL-2Rβ while avoiding signaling in bystander T cells and NK cells.”
This might turn out to be a neat system if there is sufficient clinical activity in the approach beyond what we have seen in preclinical Raji models and NHPs:
“The toxicity of STK-009 was evaluated in a non-human primate dose-escalation study. Subcutaneous administration STK-009 at anticipated therapeutic doses showed no evidence of toxicity or biological effect on immune cells expressing the wild-type IL-2 receptor. Pharmacokinetic analysis of STK-009 in this study showed stable exposure with minimal clearance, demonstrating the selectivity of STK-009.”
Comment: There is plenty of research to go yet but before anyone scoffs a preclinical asset might too early to consider, remember SynthorX and their not alpha IL-2, which Sanofi picked up (clinical data sight unseen) for a pretty penny. Whether this particular approach improves the efficacy and durability of CAR T cell therapies for patients remains to be demonstrated, but definitely an new concept to watch out for — Renier Brentjens idea of armoured CARs with secreting cytokine functions might well be coming to clinical trials a lot sooner than many observers realise!
4. Mechanisms of resistance
This is a particularly interesting area of research for me because it is becoming apparent that the mechanisms of resistance or immune escape are quite different when looking at say, CD19 versus BCMA CARs.
CD19 has a high proportion – perhaps one third of patients – who develop resistance and disease recurrence due to antigen loss, which means the CAR-T cells can no longer ‘see’ the protein target on the leukemia cells. In contrast, antigen loss is much rarer (< 5%) with BCMA-directed constructs. We don’t know why, CD19 just seems more susceptible in this fashion. In this respect, BCMA is much more like CD20, which is also less prone to antigen downregulation.
Researchers in both industry and academic are therefore exploring different mechanisms of resistance in multiple myeloma with regards to BCMA.
One relevant abstract this year on this very topic from Dr Mehmet Samur and colleagues at Dana Farber and BMS/bluebird looks at biallelic loss of BCMA in patients who received ide-cel (bb2121) in a small sample of patients who experienced relapse:
“Soluble BCMA level (produced predominantly by MM cells) was high before the first CAR T cell infusion and dropped significantly to a very low level coinciding with the clinical response; however, it remained low even at the time of relapse with increase burden of MM, indicating a lack of BCMA production by MM cells.”
Were there any genomic changes of relevance? Whole exome sequencing (WES) was employed to look at 16p and 17p deletions:
“Before first CAR T cell infusion, 4% MM cells showed deletion 17p, while after second infusion both WES and scRNAseq prediction showed that del17p and del16p were clonal, and longitudinal scRNAseq analysis indicated that del17p and del16p co-occurred in the same clone.”
There’s also more information to be gleaned from the analysis:
“WES identified a subclonal nonsense mutation (p.Q38 ) in BCMA that creates an early stop codon in the BCMA gene. This biallelic BCMA deletion, acquired with one copy loss and a 2nd loss-of-function mutation, provides the molecular basis for lack of BCMA expression in MM cells at the time of relapse. Our data showed that both TP53 and BCMA had deletion in one allele and mutation in the second allele.”
Comment: What’s scary about these data is they highlight the ability of the MM cells to survive without BCMA expression, meaning there’s multiple oncogenic drivers at play. This makes it a rather tricky target to consider in the context of overcoming resistance, never mind the increasing number of BCMA CARs coming along down the pike in the wake of bluebird/BMS.
Meanwhile, Terry Fry’s group have long been working on enhancements to their CD22 CAR (see earlier mention from 2012!) – initial results were quite disappointing considering CD22 upregulation was identified as a consequence of CD19 antigen loss with tiso-cel and other CD19-based CARs.
This time around they have gone back to basics to reconfigure the design and enhance antigen sensitivity of the CD22 CAR via modulation of the affinity and linker-length of the scFV backbone. Does this help performance – after all, previous clinical experience has demonstrated the importance of using a short linker (consisting of a single G4S sequence) between the heavy and light chains of the m971 scFv.
New CD22 CARs were generated with either a short or long linker and evaluated in vitro and in vivo for optimal performance:
“While the short linker improved proliferation in vitro, there was no significant impact of linker length on cytokine production or lysis of CD22Lo NALM6. In a xenograft model, HA-CD22 CAR T cells with the long-linker demonstrated slower progression of CD22Lo leukemia and significantly prolonged survival of NSG mice with CD22WT leukemia relative to HA-CD22 CAR T cells with the short-linker (p < 0.01).”
Comment: The researchers were of the opinion that increasing the affinity of the scFv is a promising strategy for enhancing CAR sensitivity to low levels of target antigen – with the potential to decrease post-CAR T cell relapses due to antigen downregulation. Next steps are to evaluate the new construct in the clinic and determine if the design changes have a major impact on patient outcomes in CD19-relapsed disease.
5. Biomarkers and intriguing translational work
a) Markers of response
The main body of work here which should be interesting comes from the BMS biomarker analysis mentioned earlier, plus some updates on the work from the Mackall lab on their CD59+ T cell population findings.
One abstract in particular by Dr Robbie Majzner and his Stanford colleagues explores how CD58 impacts the durable responses to CD19 axi-cel therapy in large B cell lymphomas (LBCL). What’s really interesting is the defects can be overcome with some nifty engineering changes to the CAR construct.
So why study CD58 and what does it do?
“Mutations in and loss of expression of LFA–3 (CD58) have been described in approximately 20% of cases of LBCL. As the ligand for CD2 on T cells, CD58 provides costimulation to T cells and CD58 loss or mutation has been linked to immune resistance in LBCL.”
In other words, those patients with intact CD58 expression have good outcomes (durable complete responses) and those who experience mutational changes have suboptimal response (partial or no responses) – the latter group are most at risk of early relapse in the first 6 months on the Kaplan-Meier curves.
The group thus sought address the latter issue by looking closely at the underlying biology:
“CD2, the T cell ligand for CD58, plays both an adhesive role and a costimulatory role in T cells. CD2 knockout resulted in significantly reduced cytokine production after CAR stimulation. Re-expression of only the CD2 extracellular domain did not rescue CAR function, indicating that CD2 signaling is essential for full CAR activation.
Additionally, when we stimulated CD19 CAR T cells with anti-idiotype antibody (CAR stimulation), soluble CD58 (CD2 stimulation), or both, we observed significantly enhanced phosphorylation of both CD3ζ and ERK by western blot in CAR T cells stimulated through both the CAR and CD2.”
What does this mean in terms of practical CAR design?
“To overcome CD58 loss in LBCL, we generated second- and third-generation CAR T cell constructs integrating CD2 costimulatory domains within the CAR molecule. While these cis constructs demonstrated increased potency against CD58KO cells in vitro, they were unable to ultimately overcome CD58 loss in vivo. However, when CARs were co-expressed with an additional CD2 receptor in trans, they mediated significant anti-tumor activity in vivo, overcoming CD58 knockout in tumor cells.”
Comment: This elegant work clearly demonstrates that CD58 status is an important biomarker for durable response to CAR T cells in LBCL. They were able to generate address this issue with CAR T cells capable of overcoming CD58 loss in B cell malignancies. The significance of this work is potentially patients could be evaluated up front to determine if their CD58 status was intact or mutated and a CD19 CAR selected as appropriate for optimal response – pretty cool if this turns out to be a useful strategy for improving the number of durable at 6 months, thereby reducing the early attrition rate on the survival curve!
b) Markers of decreased response
On the other side of the coin, are there useful indicators of where patients might do less well in response to CAR-T cell therapy? If there are, then could these markers be used to determine where potential candidates might fare better with alternative approaches, such as T cell engagers, for example?
A couple of things which jump to mind in terms of what can impact outcomes from the get-go are prior treatments and any lingering side effects from such therapies going into a CAR-T regimen. Let’s look next at an example for each case…
i) Prior therapies having a negative impact on subsequent CAR-T responses
This one might well be obvious – it’s something we’ve argued in the past – but if you consider giving say, a CD19 antibody such as blinatumomab then the target could (and very likely is) be downregulated as immune escape mechanisms lead to antigen loss. The Novartis tiso-cel trials wise excluded patients who had received prior CD19 based therapies.
In the real world it’s a different matter entirely – so now you’re thinking about a referring such patients to a tertiary centre for consideration of CD19 CAR T cell therapy such as tiso-cel – is this a good idea? No. This is where I would argue most strongly for a different target such as a CD20-based therapy, since you’re aiming to attack the cancer from a very different angle.
The debate has raged on since the patients were excluded from the trials – I’ve actually heard a few Community Hem/Oncs at tiso-cel presentations mumble about ‘well, we don’t know for sure’ on the other side of poster boards and managed to resist rolling my eyes or sighing at such nonsense and seriously hoped no one would do a clinical trial to determine the obvious!
Sure enough, some researchers have finally looked at the clinical evidence for answers in a retrospective analysis of 420 patients from 7 centres. Here’s the crux of the findings:
“Seventy-five (17.8%) patients received blina prior to CD19 CAR. The median time from last blina use to CD19 CAR was 129 days (IQR, 79–304 days). Blina was associated with an increased risk of CAR non-response; 13/71 (18.3%) blina patients versus 24/341 (7.0%) non-blina patients were non-responders (p=0.0052).
Ten of 71 (14.1%) were non-responders to both CD19 CAR and blina; 19 of 29 (66%) blina non-responders achieved remission with subsequent CD19 CAR.”
In short, they demonstrated a clear association with blina use and
- Increased risk of CAR non-response
- Worse RFS and EFS
- A trend towards a higher incidence of pre-CAR CD19 dim disease.
Comment: Case closed – sequencing these CD19 therapies is not a good idea in theory or in clinical practice and may even be detrimental because the patients have been denied the opportunity for better outcomes with a different approach such as CD20-based regimens.
ii) Side effects from prior therapies having a negative impact on CAR-T outcomes
In this study, Seattle researchers sought to “determine the rate of clinically significant cytopenias and identify pre- and post- treatment factors associated with cytopenias after axi-cel.”
In all, over 50 patients (41 DLBCL, 1 PMBCL, 11 tFL) received axi-cel in the study with one third (36%) having bulky disease (> 5 cm) while nearly half (43%) had elevated LDH (>210 U/L). One third (32%) received a prior SCT (16 autologous, 1 allogeneic) before CAR-T therapy and approx. half (49%) received bridging therapy between leucopheresis and lymphodepletion.
Grade 3+ CRS and neurotoxicity was observed in 11% and 15% patients, respectively. The majority (79%) patients had severe cytopenia.
In all, the authors concluded:
“Severe cytopenia is common after axi-cel, especially in non-responders in need of subsequent therapies. Our detailed analysis of pre- and post- CAR-T variables identified pre-CAR-T cytopenia and transfusion needs as well as post-CAR-T steroid use as potential predictors of day 30 severe cytopenia in patients with LCL receiving axi-cel.”
Comment: This is a useful clinical finding because if the cytopenias are indicative of poor response then more aggressive clinical management protocl can be devised to optimse treatment earlier rather than later. What may follow is further research to determine what drives the cytopenias happening in the first place? Are there risk factors involved which could predict the incidence for prophylactic intervention and changing the subsequent negative outcomes?
iii) Impact of the microbiome on outcomes
This analysis should not be surprise after all the work with immune checkpoint blockade published over the last five years with both the microbiome and antibiotics!
Here, Dr Viktoria Blumenberg and colleagues show clearly how both antibiotics and low gut microbiome diversity can have a negative impact on CAR-T cell therapy performance in terms of both decreased response rates and also higher toxicities in B cell pediatric ALL and adult DLBCL patients. They adminstered either tiso-cell, liso-cel, or in-house manufactured CD19-targeted CD28–4–1BB-CD3ζ CAR T cells to their patients (n=60 for the anti-infective analysis) and collected faecal specimens from 33 of them for the microbiome assessment.
The found the following:
- Loss of diversity correlated significantly with the occurrence of CRS (p < 0.001, univariate GLM with Shannon diversity index vs. occurrence of CRS, irrespective of grade).
- Diversity loss was not associated with clinical response at day 90 or the development of ICANS.
- Expansion of enterococci was found in samples of patients who received antibiotic treatment.
- Patients receiving anti-infectives up to two weeks prior to CAR T cell transfusion display a significantly lower response rate compared to patients who have been treated with antibiotic and / or antimycotic treatment after day 0.
Comment: The authors concluded “the assessment of the gut microbial taxa of CAR T cell patients might serve as a predictive biomarker for immunotoxicity and, eventually, treatment response.” These results are not at all surprising given the previous findings from resarchers such as Tom Gajewski and Marcel van den Brink, which we first covered five years ago! Aside from the timing of antibiotics having a negative impact, we can see a similar trend with steroid use too, as UK researchers found out to their detriment – steroids have long been known to affect T cell activation from solid organ transplantation with dexamethsaone and methotrexate and can dampen the immune response very effectively indeed!
c) Novel imaging findings
Dr Crystall Mackall’s lab have certainly been prolific on several fronts of late. This time around they showcase some really useful research based on molecular imaging with immunoPET to non-invasively visualise CAR T cells in vivo. Using this approach they could determine exactly where do the CAR-T cells go in the body? In the mice, I was surprised to see they could trace their presence to bone, spleen, and even the bone marrow, for example.
The data from patients (n=31) receiving axicabtagene ciloleucel (axi-cel) for R/R DLBCL was analysed using mass spectrometry (CyTOF), along with mice treated with the CAR. They then looked at the ex vivo ICOS expression on human and murine CD19–28z CAR T cells present in blood. Essentially ICOS becomes an activation marker whose transcription is upregulated and sustained during in vitro culture:
“ICOS was preferentially expressed on CAR+ T cells recovered at day 7 from axi-cel treated patients compared with CAR cells (p<0.001)”
Overall, the immunoPET approach was able to monitor the in vivo dynamics of CAR T cell migration, expansion, and persistence, which did not require the addition of clunkier current approaches using reporter genes or ex vivo labelling. This is a really useful finding, which means it will be applicable to the clinic for the study of any commercially available and investigational CAR T cell products. Expect to see studies evolve using this new approach.
6. Adverse events
Well, let’s be honest – it’s not all rose tinted glasses in any clinical research endeavour and side effects are to be expected. The issues whether they can be predicted, are manageable (prophylactically or therapeutically), or are lethal and problematic.
a) Allogene
See earlier discussion on CD52 lymphodepletion and increased risk of infectious complications.
b) Bleeding and thrombosis with endothelial cell dysfunction
This is another useful analysis from adult patients with R/R DLBCL or ALL who received either axi-cel (n=90) or a bispecific CD 19/22 CAR construct (N=40) based on CD28 and 4–1BB costimulatory domains, respectively.
We have heard much about CRS and neurotoxicites, but what about life-threatening consumptive coagulopathies? As Johnsrud and colleagues pointed out, “the underlying pathobiology of such hemostatic defects and their distinct clinical sequelae remains obscure.”
Platelet counts and coagulation parameters were collected prior to lymphodepletion then on days 0, 3, 7, 14, 21, 28, 60 and 90.
The number of events in this sample were not large – less than 10% in each case: “12 (9.2%) and 8 (6.2%) patients developed bleeding and thrombotic complications in the first three months after CAR-T infusion, respectively.”
There were some emergent patterns of relevance identified:
- All bleeding events occurred between days 0–30 (median 17.5, range 8–30)
- Thrombotic events occurred between days 2–91 (median day 29, range, 2–91)
- Two (1.5%) patients experienced both bleeding and thrombosis
- Bleeding events coincided with the onset of thrombocytopenia and hypofibrinogenemia
- Patients who bled had lower platelet (median 22.5 vs. 47 K/uL; p=0.03) and fibrinogen (median 151 vs. 351 ug/mL; p=0.007) nadirs in the first 30 days compared to those without bleeding.
Now here’s where things get intriguing because in addition, neurotox was found to be associated with PT prolongation, hypofibrinogenemia, and increased fibrin degradation, in addition to both bleeding and thrombosis. This broader pattern led researchers to conclude:
“These results suggest that a systemic coagulopathy coincides with high grade ICANS and whether these neurologic events truly represent sequelae of widespread vascular dysfunction warrants further investigation.”
Comment: I’m left pondering whether any obvious risk factors for bleeding and thrombotic complications, but the researchers stated further prospective study is required to determine these. Overall, this an important piece of work with practical implications for clinical management and monitoring.
And Finally…
There are a couple of additional studies to highlight at ASH, which don’t really fit in any of the categories selected above in this Preview.
Instead of autologous (self cells) or allogeneic (healthy donors), there’s also human induced pluripotent stem cells (iPSCs), which can be used to make engineered T cells.
Several companies are exploring this approach including Takeda and also Fate Therapeutics. The latter have been expanding their novel iPSC derived platform with several T and NK cell therapies being evaluated both preclinically and clinically.
There was an update on Fate’s FT819 at this year’s ASH meeting. This is an off-the-shelf CD19 based CAR T cell therapy:
“The ability to consistently administer more than a single dose of CAR19 T cells enables dosing schedules that may reduce the risk of potentially life-threatening toxicities such as cytokine release syndrome and neurotoxicity, while maintaining or improving the depth and durability of anti-tumor responses.”
They describe the construct design as thus:
“FT819 is engineered with the following features designed to improve the safety and efficacy of CAR T cell therapy: a novel 1XX CAR signaling domain, which has been shown to extend T cell effector function without eliciting exhaustion; integration of the CAR transgene directly into the T cell receptor alpha constant (TRAC) locus, which has been shown to promote uniform CAR expression and enhanced T cell potency; and complete bi-allelic disruption of T cell receptor (TCR) expression for the prevention of graft-versus-host disease (GVHD).”
Other targets of interest to watch out for:
Beyond CD19, CD20, CD22, BCMA, GPCR5D, CD123, and various other targets, there’s also CD33 and CD38 to think about in AML and myeloma, respectively.
Vor BioPharma recently announced their deal with the NCI for a CD33 directed CAR-T cell therapy developed by Dr Terry Fry’s lab. We interviewed their CEO, Dr Robert Ang, at last year’s meeting for additional context around their strategic intent in the CD33 space.
Meanwhile, a Chinese group have now developed and conducted initial testing in 6 R/R AML patients post allo HSCT on a CD38-directed construct. It’s early days with this idea but a decent start, nevertheless.
My last selection is a dual bispecific CAR against BCMA and CD19, which is a FasTCAR from Gracell being evaluated in multiple myeloma. They have, however, been guilty in the past of submitting the same data to several conferences and have avoided discussion about their platform and research, which doesn’t help their case in being widely accepted.
This latest construct basically consists of linked BCMA and CD19 scFvs, joined by a CD8 hinge, transmembrane domain, co-stimulatory domain, and CD3z.
At ASH20 they offer initial data in 16 heavily pre-treated myeloma patients, although this may be updated with more people in the actual presentation:
- 15 out of 16 patients responded to treatment (ORR 93.8%) in all dose levels with the earliest response observed at day 28.
- Best response to date is MRD- CR/sCR in 9/16 patients (56.3%).
- In DL3 100% (6/6) of patients achieved sCR, 3 at data cut off had been confirmed by PET-CT.
- In all response evaluable patients, 78.6% (11/14) were MRD- by flow at month 1, and 100% at month 3 (11/11) and 6 (10/10).
Comment: Of course, it’s early days yet and many people are somewhat sceptical about Chinese claims for faster manufacturing and processes, never mind innovative design ideas leading to dramatic responses in a small sample size. That said, an 93.8% ORR to date is not to be sniffed at or dismissed so we await the presentation to learn more about their research.
In the next ASH Preview ahead of the meeting, we’ll be taking a look at a very different topic — what’s hot in small molecule drug development in hematologic maligancies…
[/restrict