
On yer bike!
Today’s post features an intriguing story which brings together some science around oncogenes, solid tumours, NK cells, T cells, and the immune system – what’s not to like?
What if we want to improve the response to checkpoint blockade in solid tumours? There are many ways to go about achieving this, much like choosing a bike from a rack of available selections.
Of course they will each have pros and cons and need to be fit for purpose, but first we need to think about some of the underlying biology driving the lack of responses and potential solutions to fix them.
In this article, we highlight some useful new research pointing the way for intrepid researchers and companies seeking to address the critical issues…
BSB subscribers can read our latest oncology review plus commentary and analysis – you can either log-in or click to access.
This content is restricted to subscribers
Background
It’s time to talk about tackling melanoma – and more specifically, how can we increase the number of long term responders to checkpoint blockade?
One of the many challenges with both small molecule approaches and immunotherapy is we might be focused on certain targets while ignoring many others in the equation… usually because a company either doesn’t have a particular therapeutic in their pipeline or it’s one of the intractable targets such as MYC, TP53/p53, KRAS, β-catenin etc.
Consider this relevant point made by Benjamin and colleagues way back in 2008:
“The p53 tumor suppressor gene and gene product are among the most diverse and complex molecules involved in cellular functions. Genetic alterations within the p53 gene have been shown to have a direct correlation with cancer development and have been shown to occur in nearly 50% of all cancers. p53 mutations are particularly common in skin cancers and UV irradiation has been shown to be a primary cause of specific ‘signature’ mutations that can result in oncogenic transformation.”
A pertinent issue with tumour suppressors is they involve loss of function, which means we are essentially faced with two potential strategies to induce apoptosis of the cancer cells – either restore normal function or use a synthetic lethal approach.
Indeed the researchers went on to hint at the latter with their mention of DNA damage:
“Activation of p53 tumor suppressor protein occurs in response to variety of cellular stresses including DNA damage, oncogenic stimulation, hypoxia, oxidative stress or telomere shortening and directs cells toward cell cycle arrest or apoptosis depending on the amount of DNA damage.”
A year later, Len Zon’s lab conducted some elegant experiments and determined how we start to get at the heart of the issue:
“Oncogenic NRAS cooperates with p53 loss to generate melanoma in zebrafish.”
“Experimentally, oncogenic BRAF or HRAS is capable of contributing to melanoma, but only in the context of co-operating loss-of-function mutations in p53, INK4, and/or ARF tumor suppressors.”
In other words, if we ignore the NRAS and p53 loss then melanoma is still being driven by critical oncogenes and any other therapeutic intervention is thus likely to have limited durability in many cases.
More recently there have been a couple of helpful studies linking p53 to acquired resistance to therapy in melanoma:
- Potential for targeting p53 to overcome acquired MAPKi-resistant melanoma (link).
- Targeting p53 for melanoma treatment: counteracting tumour proliferation, dissemination and therapeutic resistance (link).
The role of ill-fated MDM2 inhibitors such as RG7112 and others in addressing p53 was, however, doomed by too a narrow therapeutic window, which means we clearly needed to start thinking about alternative novel approaches. Amgen’s AMG 232 is still going, although there are no guarantees it will break the consistent MDM2 trend observed to date.
Another critical question is exactly how does p53 loss relate to the immune system performance?
After all, I’ve often heard cancer immunologists proclaim the immune system simply doesn’t care about mutations or various oncogenic aberrations because the more you have, the more likely you are to respond if the tumour burden is higher… Except this sweeping and broad generalisation isn’t entirely true.
We have seen, for example, the negative effects of other tumour suppressors cropping up over time such as PTEN whereby they are quite capable of inducing immune escape, and ultimately, loss of response to checkpoint therapy. In addition, Gerard Evan (Cambridge) has clearly demonstrated MYC and KRAS can cooperate together to drive out T cells, creating an immune desert where there will be little hope of seeing a response with checkpoint blockade. Imagine what might happen if we added some tumour suppressors into the mix like PTEN? We know from Dana Farber’s case study at ASCO a few years ago it can lead to sudden loss of response to pembrolizumab and disease recurrence.
Does the same thing happen with p53 loss, and if so, would activating p53 have a positive impact? It turns out the answer may well be yes, according to recent research published in Cancer Discovery:
- Pharmacologic activation of p53 triggers viral mimicry response thereby abolishing tumour immune evasion and promoting antitumor immunity (link).
You start to get the idea now that finding a therapeutic approach to target p53 might be a useful strategy to pursue in melanoma, especially if one of the hostile elements in the milieu can be tamed somewhat. So how might we go about doing this?

Prof Nick Huntington via Zoom in 2020
Yesterday, there was a new paper out jointly from labs at Penn State (Gavin Robertson) and Monash (Nick Huntington, right), which speaks to this very issue.
Dinavahi and colleagues explored how targeting WEE1/AKT restores p53-dependent NK cell activation to induce immune checkpoint blockade responses in ‘cold’ melanoma, so let’s look at their findings and what they mean…
What do we learn from the latest research?
Let’s pull together a series of key research in this area and start with Dinavahi et al., (2022), who noted an important background finding:
“Approximately 80% of melanomas have wildtype p53, with high MDM2/4 proteins leading to the functional inactivation of the p53 pathway. The MDM proteins reduce the transcriptional transactivation of proapoptotic proteins in tumor cells and promote an immune-suppressive TME.”
We have already mentioned the toxicity of the MDM2 class of inhibitors, so are there alternative approaches which can be considered?
“One alternative is to indirectly increase p53-mediated transcriptional activity in tumor cells and reverse the immunosuppressive TME by targeting AKT/WEE1 pathways. This approach synergistically increased p53.”
To this end they decided to explore the potential for small molecule inhibition against these targets using adavosertib (WEE1) and capivasertib (AKT), both from AstraZeneca, in relevant cell line experiments.
Here is where things get interesting – consider this particular finding:
WEE1/AKT inhibition leads to p53-dependent expression of ligands to activating NK cell receptors
They go on to note:
“Reports indicate that the p53 pathway is important for upregulation of NKG2D ligands, such as ULBP1 and ULBP2, leading to increased tumor recognition and destruction. Furthermore, p53 activity within NK cells has been shown to be critical for functional maturation.”
One of the cited references for the first statement was from Textor and colleagues (2011) who noted:
“… induction of wild-type p53, but not mutant p53, strongly upregulated mRNA and cell surface expression of ULBP1 and –2, whereas expression of other NK cell ligands was not affected.”
Additionally in the last statement, the source of this finding was Collin et al., (2017), who also pointed out the importance of NK cells from their work validating Trp53 as a gene regulating the functional maturation of NK cells:
“Natural killer cells constitute potent innate lymphoid cells that play a major role in both tumor immunosurveillance and viral clearance via their effector functions.”
By the way, if you’ve ever wondered why elephants have a low incidence of cancer, the authors noted these creatures have 20 copies of Trp53, which may mean they have well tuned immunosurveillance mechanisms in place to ensure protection!
While we have seen considerable efforts expended on PD-L1 and tumour mutation burden (TMB) as potential predictors of response to immunotherapy, people are gradually beginning to look outside the T cell focus at other immune cells, including NK cells. Do they have much impact in this context?
You betcha, as the researchers highlight from Pahl and Cerwenka’s (2017) review:
“Despite their low abundance compared to CD8+ T cells within most tumor types, high intra-tumoral NK cell frequency correlates with improved overall survival in melanoma, lung, head and neck, gastric, and colorectal carcinomas, underscoring their role in immunosurveillance.”
One of the ongoing challenges with T cells is they need to recognise tumour associated antigens in order to function, but effectively become blind when the target is downregulated or lost, while NK cells recognise tumour cells with the activating receptor NKG2D. So how does this relate to p53?
“Emerging evidence suggests that restoration of p53 signaling within tumor cells might promote NK cell recruitment, activation, and function in the tumor microenvironment.”
Dinavahi and colleagues thus decided to evaluate the efficacy of modulating the p53 pathway in cancer cells containing wild type TP53 (where the pathway is inactivated by MDM protein overexpression) to induce NK cell-driven immunotherapy responses.
Here’s what they observed:
“Pharmacological inhibition of AKT and WEE1 using capivasertib and adavosertib, respectively, led to significant synergistic inhibition of melanoma cell growth and increased calreticulin signals indicative of immunogenic cell death.
Increase in immunogenic cell death further increased NK cell ligand expression on melanoma and enhanced recognition, leading to NK cell activation and cytotoxicity. Combining p53 pathway activation with anti-PD–1 immunotherapy increased NK cell, cDC1, and CD8+ T cell recruitment into the TME, without toxicity, and led to regression of syngeneic melanomas, which are typically anti-PD–1–insensitive.
Depletion of NK cells, CD8+ T cells, or inactivation of p53 in melanoma cells ablated the therapeutic efficacy, thus, validating the importance of the p53 pathway in NK cell-based potentiation of anti-PD–1 immunotherapy responses.”
In other words, using WEE1 or AKT inhibition might be a useful approach to restoring p53 function in wild type TP53 melanomas which have not responded to checkpoint blockade with the potential to induce fewer toxicities than seen with MDM2 inhibitors in the past. Of course, these agents are not without adverse events themselves, but neither approach has the sheer severity of myelosuppressive events seen with targeting MDM2.
Overall, the scientists concluded:
“Activation of the p53 pathway can increase apoptosis of cancer cells, leading to enhanced antigen presentation, and can stimulate natural killer cells through expression of stress ligands. Therefore, modulation of the p53 pathway in cancer cells with wildtype TP53 has the potential to enhance tumor immunogenicity to NK cells, produce an inflammatory TME, and ultimately lead to tumor regression.”
Additional Commentary
There are a few ongoing trials evaluating DDR agents in combination with checkpoint blockade. Last year Sun et al., looked at 92 trials in this context, and noticed PARP inhibitors dominated (84%), while WEE1 inhibitor studies were in the minority (2%):
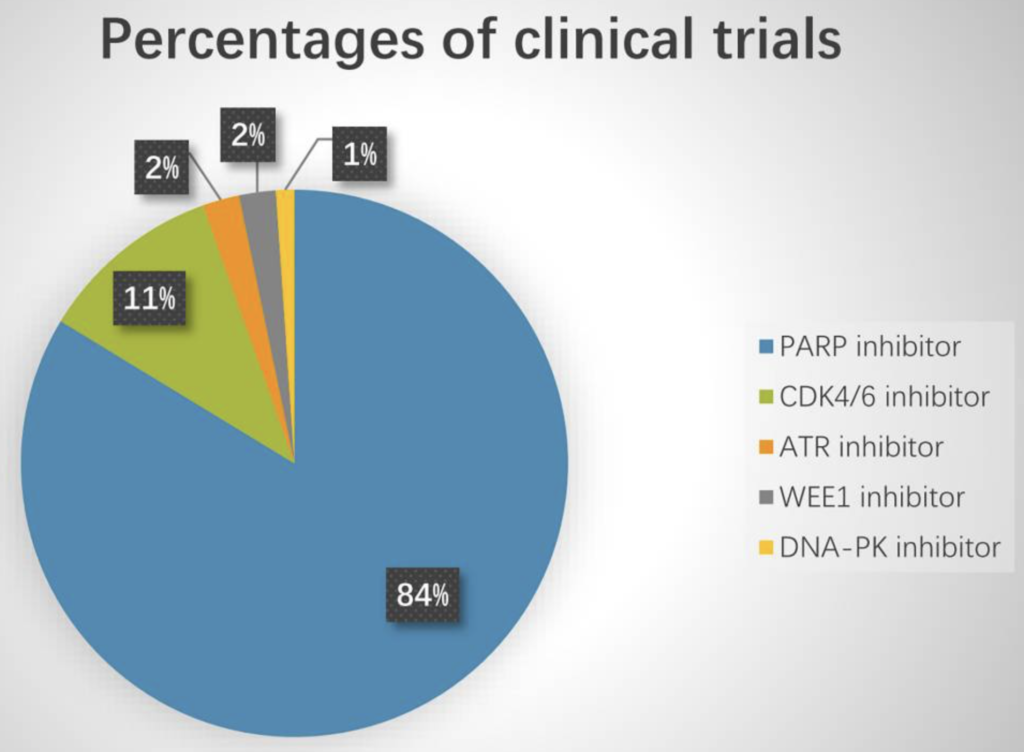
Source: Sun et al., (2021)
A phase 1 trial with initial results at ASCO19 evaluated an adavosertib plus durvalumab combination in advanced solid tumours produced modest activity results at best (DCR 36%).
These results do make me wonder if an additional NK cell targeting agent is needed in the mix for patients with advanced cancers since the tumour burden is higher and milieu more hostile than we see in either cell lines or syngeneic models.
To this end, Innate Pharma and AstraZeneca also have monalizumab (NKG2A checkpoint inhibitor) as well as a trispecific NK cell engager (ANKET) to induce synthetic immunity by engaging activating receptors (NKp46 and CD16), a tumour antigen and an IL–2 receptor (via an IL–2 variant) in a single molecule, which they could potentially throw in the mix. Monalizumab is currently being investigated in SCCHN and NSCLC rather than melanoma, however, although it isn’t clear what they are doing with their collaboration on the ANKET yet since it is still in preclinical development.
The short video below from Innate Pharma nicely illustrates the MOA and how the trispecific might work in engaging NK and T cells in cancer:
Innate Pharma aren’t the only company developing an NK cell trispecific – there’s also Dragonfly Therapeutics and Artiva Bio to consider in this mix. The latter just announced a collaboration earlier this month with Merck to develop an approach involving NK cells with trispecific NK cell engagers, a follow-on to their deal in January to explore CAR-NK cells targeting certain solid tumour-associated antigens. Meanwhile Dragonfly have several deals with big Pharmas such as Merck, AbbVie, and BMS to develop their TriNKETs against mostly undisclosed cancer and autoimmune targets.
You can see then we are starting to see a range of NK cell approaches evolve now from CAR-NKs, plus NK cell engagers (such as Affimed) to TriKEs and TriNKETs, and now ANKETs, as well as NK memory-like cells from cord blood are in the mix.
The latest CAR-NK cell therapy approach to receive positive news was from Nkarta Therapeutics, a biotech founded by Dario Campana, a well regarded NK cell expert. Earlier this week the company announced new data on their co-lead candidates, NKX101 (NKG2D) and NKX019 (CD19), with early signs of activity in “3 of 5 patients with relapsed / refractory AML and 5 of 6 patients with relapsed / refractory NHL”, respectively. I would urge caution with both Nkarta and the Affimed/MDACC data since both have a small number of patients with limited follow-up and focus on cherry picking a tiny subset of people who received the RP2D, which may not be at all representative of responses in a much larger cohort.
While CAR-T cells therapies have seen challenges to be addressed with lethal CRS and neurotoxicity, bispecifics have been much kinder on the patient over (not quite to the extent the salacious new headlines might give us the impression re: CARs kill, BiKEs thrill though!)
Nevertheless, given the generally poor durability of NK cell therapies seen to date, take a look at the number of patients ongoing at 6 months for a much fairer reflection of the persistency of the product (see Nkarta’s swimmer plot below as an example):
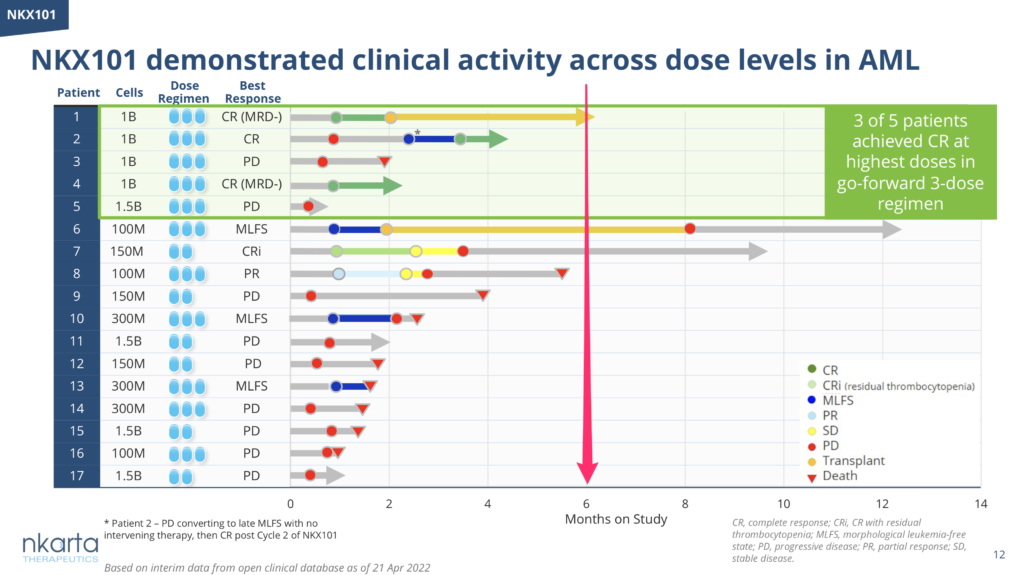
Source: Nkarta Tx
I bring this up because we already heard from Prof John Gribben (Barts) at the recent CAR-T meeting that the second cohort of patients in the Rezvani/MDACC CAR-NK trial didn’t do as well (ORR 38%) as the gaudy stats reported from the first cohort (ORR 73%) – caveat emptor. Will the same thing happen with the Nkarta CAR-NK data? We’ll have to wait and see.
Overall, it’s a promising time to be in the NK cell niche, with much work yet to come and the outcome uncertain on which approach will win out in the solid tumour setting with good durability.
And Finally…
With ASCO coming up very soon and the abstract titles dropping today, I was curious to see if there were any relevant TP53/p53 or WEE1/AKT abstracts of interest on this topic.
There wasn’t much on WEE1 or AKT:
- Do not expect too much on IMP7068 from Shanghai based Impact Therapeutics since their phase 1 trial evaluating the WEE1 inhibitor only just started enrolling patients as of March last year in the middle of the Covid19 pandemic. Wee1 is a synthetic lethal target in p53-mutant tumours.
- Meanwhile Debio Pharma’s WEE1 inhibitor (Debio 0123) has a phase 1 readout in advanced solid tumours.
Both the AstraZeneca (adavosertib) and Zentalis (ZN-c3) have been limited in terms of combinability due to myelosuppression and drug-drug interactions, respectively, although the latter did show some encouraging initial signs of activity in patients with TP53 mutations at AACR earlier this month, while 3 confirmed responses out of 55 treated inevitably led to negative stock action.
If you are going to go down the route of claiming a superior, more differentiated product due to greater selectivity (see below) then you really need to generate greater than 10% activity, rather than cherry picking a higher response rate in a tiny subset of 7 patients!
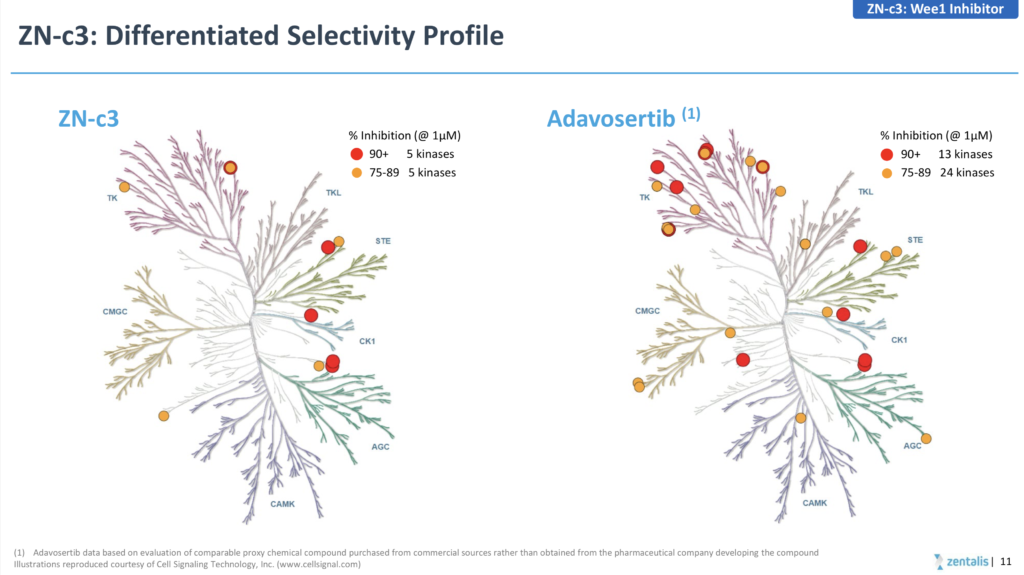
Source: Zentalis
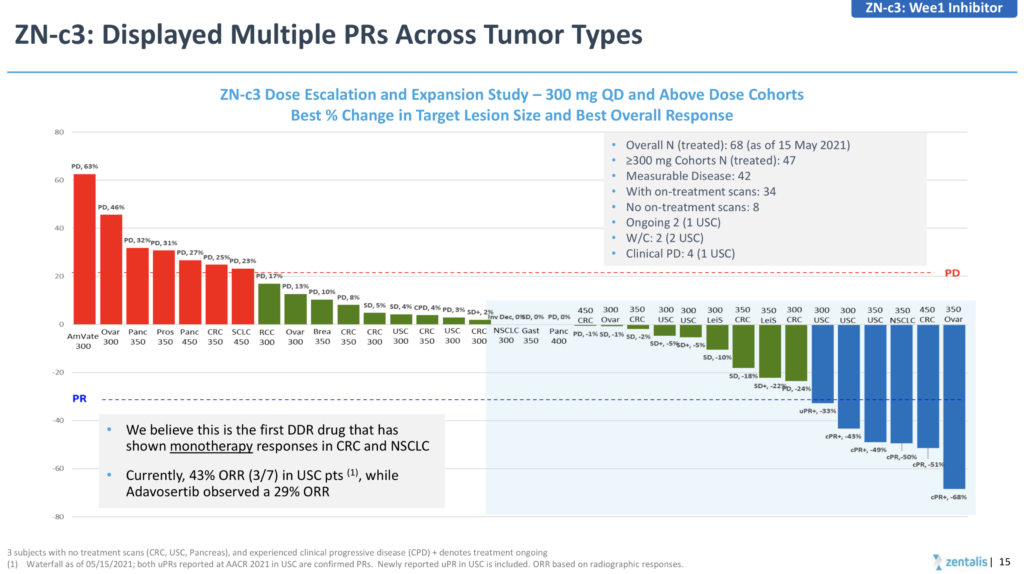
Source: Zentalis
Where AZN have a clearer edge is in the potential ability to combine adavosertib with the new selective PARP1 inhibitor (AZN-5305), which debuted at AACR with some encouraging FIH clinical data.
There’s also a couple of TP53/p53 related items at ASCO:
- In developmental therapeutics – immunotherapy there’s an abstract entitled, “Investigating the various predictive values of TP53 mutations for response to immune checkpoint inhibitors (ICIs) in different solid tumors”, which might prove to be useful.
- Amgen have new data in a poster on their MDM2 inhibitor (AMG 232, navtemadlin), in wild-type p53 soft tissue sarcoma of the extremity and body wall, a disease which has not been sensitive to immunotherapy approaches. This agent previously demonstrated a more encouraging tolerabiity profile in an early myeloma trial relative to Roche’s idasanutlin, which missed its primary endpoints in a phase 3 MIRROS trial in R/R AML, likely due in part to the narrow therapeutic window. The current STS data is in the neoadjuvant setting with concurrent radiotherapy. Recall the MOA of the MDM2 agents was based on the idea that by preventing the p53-MDM2 interaction, they enabled p53 activation, particularly in patients with TP53 wild-type status. Whether the Amgen compound will be more successful than Roche’s remains to be determined.
- Boehringer have a MDM2–p53 antagonist in early development codenamed BI 907828 and are reporting their phase 1 data in solid tumours. Again, the big question mark here is the therapeutic window so watch out for the safety profile, especially at high doses.
- A little known biotech, PMV Pharma have a small molecule (PC14586) in clinical development targeting the p53 Y220C mutation, with the aim of restoring function. They demonstrated proof of principle for this concept at AACR last year, when PC14586 was shown to convert Y220C mutant to the wildtype conformation, resulting in re-activation of p53 transcription. At ASCO next month they will be showcasing their first-in-human clinical data. I’ll be interested to see if this concept holds up and how it might compare with the Berkeley/NIBR DUBtac idea where they seek to stabilise proteins associated with loss-of-function.

 Much has been written about cytokines over the last decade in terms of various ways of employing them systematically as cancer therapeutics.
Much has been written about cytokines over the last decade in terms of various ways of employing them systematically as cancer therapeutics.
 We’ve been following this niche since NIBR scientists reported important preclinical results with their probe molecule (
We’ve been following this niche since NIBR scientists reported important preclinical results with their probe molecule ( Every now and again something intriguing comes along, which generates a flurry of interest from our readers in terms of early stage pipeline developments.
Every now and again something intriguing comes along, which generates a flurry of interest from our readers in terms of early stage pipeline developments.


